{kind=link}
Manufacturing a gene therapy with patient safety in mind
Summary
Keywords: quality standards, transfection reagents, cGMP transfection reagent, cGMP manufacturing, pharmaceuticals cGMP, medical devices cGMP, viral vectors, raw material
Pharmaceutical versus medical device cGMP manufacturing
Introduction to product quality and current Good Manufacturing Procedures (cGMP)
Current Good Manufacturing Practices (cGMP) guidelines are set by regulatory agencies and aimed at minimizing risk associated with pharmaceuticals and medical devices production by ensuring purity, safety, and efficacy of the products. In the US, it is a legal requirement of the federal government for pharmaceutical companies to comply with current cGMP set up by the Food and Drug Administration (FDA).
An industry that operates according to cGMP regulations offers products whose quality, identity, purity, and strength have been tested and confirmed according to controlled manufacturing processes. The regulations that govern cGMP require a Quality driven approach to manufacturing (often quality by design or QbD) to minimize errors, supply chain disruption, contamination, and batch failures. The goal for cGMP is to protect patients from potentially unsafe or ineffective pharmaceutical or medical devices.
In the USA, the FDA deals separately with drugs (or pharmaceuticals) and medical devices. The guidelines for pharmaceutical cGMP are documented in Title 21 Code of Federal Regulations (CFR) 210, while 21 CFR 820 describes the cGMP requirements for medical devices.
What are the differences between cGMP regulations for pharmaceutical and medical devices?
| BOX 1: Definitions of drugs (pharmaceuticals) and medical devices according to the Food, Drug and Cosmetic Act1 (21 USC 321):
Drugs “(A) articles recognized in the official United States Pharmacopoeia, official Homoeopathic Pharmacopoeia of the United States, or official National Formulary, or any supplement to any of them; and (B) articles intended for use in the diagnosis, cure, mitigation, treatment, or prevention of disease in man or other animals; and (C) articles (other than food) intended to affect the structure or any function of the body of man or other animals; and (D) articles intended for use as a component of any articles specified in clause (A), (B), or (C). . . .” (medical) device “an instrument, apparatus, implement, machine, contrivance, implant, in vitro reagent, or other similar or related article, including any component, part, or accessory, which is— (1) recognized in the official National Formulary, or the United States Pharmacopeia, or any supplement to them, (2) intended for use in the diagnosis of disease or other conditions, or in the cure, mitigation, treatment, or prevention of disease, in man or other animals, or (3) intended to affect the structure or any function of the body of man or other animals, and which does not achieve its primary intended purposes through chemical action within or on the body of man or other animals and which is not dependent upon being metabolized for the achievement of its primary intended purposes.” |
As illustrated by the definitions (Box 1), the main difference between a drug and a device is that the device is not primarily intended to induce metabolic effects on the body of man or animal, while the pharmaceutical is intended to affect the structure or the function of a body. This in turn determines the cGMP requirements for manufacturing.
Additional insights into their respective cGMP regulations may help to further understand those differences. We have compared Title 21 CFR 210 and CFR 820 for Drugs and Medical Devices respectively, and more specifically the definitions and guidelines relating to materials and specifications (Please note that the term ‘Pharmaceuticals’ is often used instead of drugs). This comparison has highlighted that whilst cGMP for medical devices put emphasis on the reproducibility of the device’s manufacturing process, cGMP for pharmaceuticals focusses on batch-to-batch reproducibility which is achieved through full validation of the manufacturing process.
In addition, cGMP for pharmaceuticals explicitly includes quality control in the manufacturing process monitored by an internal Quality Control Unit.
Finally, the definition of materials/components and product specifications is more prescriptive for cGMP for pharmaceuticals with more specific requirements compared to cGMP for medical devices (see example of materials/components below in Box 2).
| Box 2. Definitions of Materials/Components for Pharmaceuticals and Medical Devices according to Title 21 CFR 210 and CFR 820 respectively
Pharmaceuticals 210.3 Definition “In-process material means any material fabricated, compounded, blended, or derived by chemical reaction that is produced for, and used in, the preparation of the drug product.” “Component means any ingredient intended for use in the manufacture of a drug product, including those that may not appear in such drug product.” “Active ingredient means any component that is intended to furnish pharmacological activity or other direct effect in the diagnosis, cure, mitigation, treatment, or prevention of disease, or to affect the structure or any function of the body of man or other animals. The term includes those components that may undergo chemical change in the manufacture of the drug product and be present in the drug product in a modified form intended to furnish the specified activity or effect.” “Inactive ingredient means any component other than an active ingredient.” Medical Devices 820.3 Definition “Manufacturing material means any material or substance used in or used to facilitate the manufacturing process, a concomitant constituent, or a by-product constituent produced during the manufacturing process, which is present in or on the finished device as a residue or impurity not by design or intent of the manufacturer.” |
How does this relate to transfection reagents?
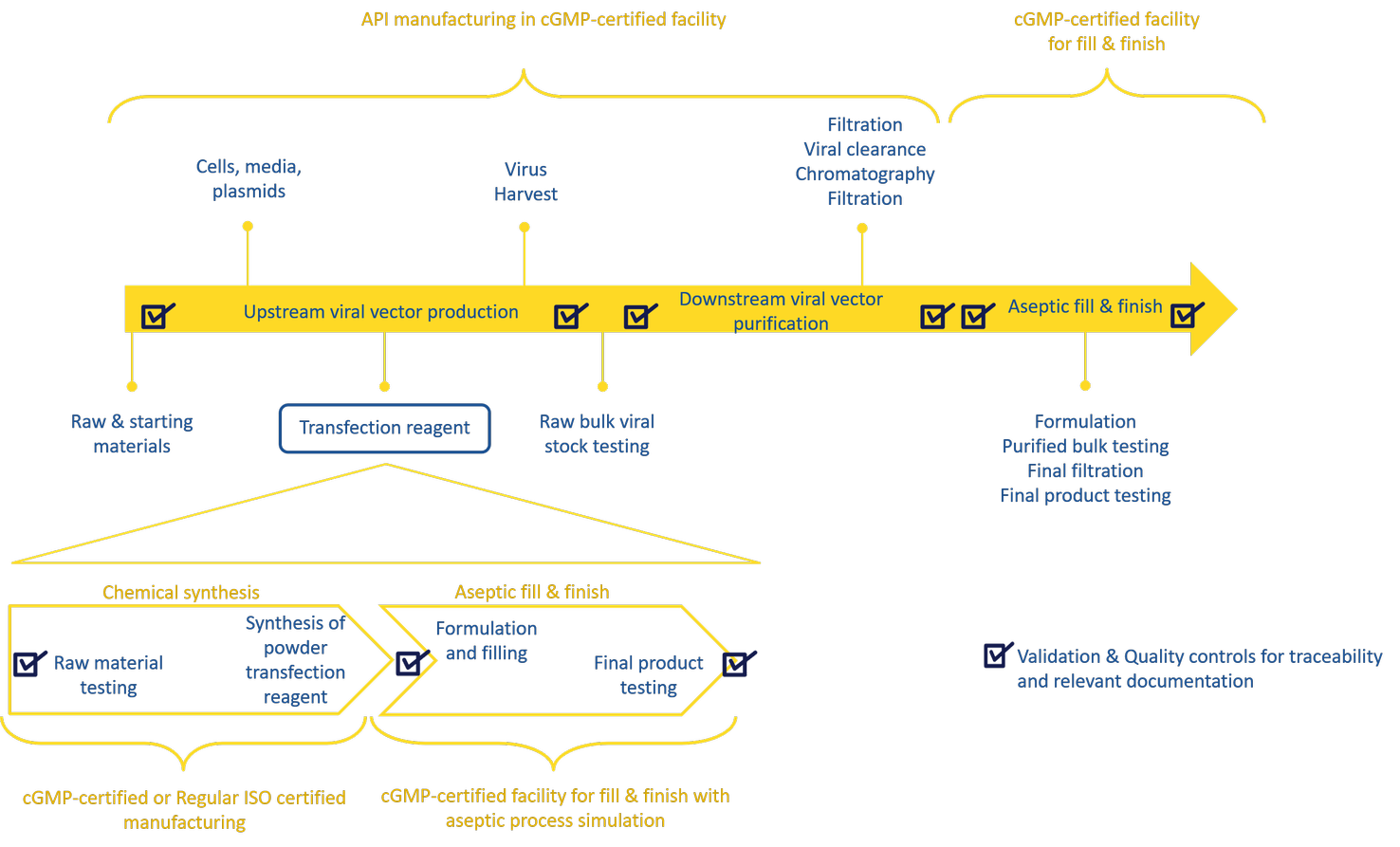
The cGMP manufacturing of transfection reagents has been growing exponentially as it has paralleled the growing demand for cGMP viral vector manufacturing for in vivo and ex-vivo gene therapy. Production of clinical grade viral vectors requires manufacturing under cGMP regulations to ensure patient safety. To do so, manufactures need to source qualified raw materials and to manage the supply chain.
As part of the risk-based approach used by viral vector developers, the availability and supply of raw and starting materials is assessed by mapping the materials and steps that are critical to mitigate the risk of manufacturing failure. Transfection reagents are not just considered as raw materials, but they are considered as critical raw material for large scale production of viral vectors. Therefore, having a reliable source of qualified transfection reagent will increase consistency of the process by improving the reproducibility of the transfection step. This will not only satisfy clinical demand, but also quality and regulatory requirements, as the critical attributes of the final product need to be validated.
Viral vector manufacturers must also mitigate additional risks which can drastically impact product release. For example, they need to minimize aspects relating to raw materials such as lack of traceability, non-controlled or un-notified manufacturing changes or failed batches. (For more details on raw materials in viral vector production, read The importance of raw materials in viral vector manufacturing: Focus on transfection reagent. Therefore, viral vector developers and manufacturers need to scrutinise how their raw materials are manufactured and check which cGMP standards they are compliant with.
Get to know your transfection reagents: what cGMP guidelines are they compliant with?
cGMP grade transfection reagents should be produced and formulated following strict guidelines. Transfection reagents are manufactured in two phases:
- Phase 1, which comprises their chemical synthesis and can be performed under cGMP
- Phase 2, which completes the process with formulation and aseptic fill & finish of the final product. It usually takes place in a cGMP-certified facility with aseptic simulation. (For details for this manufacturing process, please refer to Figure 1 of The importance of raw materials in viral vector manufacturing: Focus on transfection reagent.)
Some suppliers manufacture transfection reagents according to medical devices cGMP standards which makes the reagents suitable for viral vector manufacturing in the United States but not suitable for direct administration into humans. Those are often labelled as “for research use only and further manufacturing. Not for use in humans or animals”. Others comply to pharmaceuticals cGMP guidelines, making the transfection reagent suitable for viral vector manufacturing as well as safe for administration in humans.
Suppliers of transfection reagents produced under pharmaceuticals cGMP comply with pharmaceutical guidelines from start to finish, meeting more prescriptive product manufacturing and specifications. The use of pharmaceutical cGMP grade transfection reagents also ensures batch-to-batch consistency, traceability, and product specifications (identity, purity, potency, safety, and quality). Consistent with pharmaceutical cGMP regulations, a “full documentation” is generated with every single batch of transfection reagent to support the batch release by the Quality Assurance department. This documentation includes detailed batch production, information relating to starting material, equipment, training of personnel, process, cleaning, and of course quality controls related to the batch; ensuring batch compliance.
So, what are the advantages of using a transfection reagent manufactured under pharmaceutical cGMP over a transfection reagent manufactured under medical devices cGMP?
- Batch-to-batch consistency, as pharmaceutical cGMP compliance emphasizes reproducibility of the product and therefore it is very strict on consistency.
- Better monitoring of product quality throughout the entire manufacturing process, ensuring full process validation and traceability.
- Improved risk mitigation strategy, by having full validation and full traceability of the manufacturing process.
- Peace of mind that comes with the use of qualified raw material for the transfection step which is critical in the production of therapeutic viral vectors.
In summary, pharmaceutical cGMP grade manufacturing ensures full process validation, full traceability, and improved batch-to-batch consistency. These guidelines apply to the manufacturing process of the viral vectors but preferably also to critical raw materials used in the process such as transfection reagents in order to meet safety criteria. Therefore, our recommendation to early developers needing viral vectors, is to keep patient safety in mind throughout the development of their processes. This starts with the quality and safety of the raw materials to be used and careful selection scrutinised manufacturing subcontractors. This approach will also improve risk mitigation strategies.
(To receive the next articles directly in your mailbox, subscribe to our cGMP VIP list)
Learn more