PEIpro®
PEIpro® is the leading PEI-based DNA transfection reagent that offers flexibility and scalability for viral vector manufacturing.
![]()
![]()
Gene therapy, tissue engineering and cell therapy are quickly becoming the biggest innovations in medicine. These promising biotechnologies aim to correct, restore or repair diseased tissues and function and represent a major shift from classical drug-based therapies. For this reason, regulators worldwide have developed completely new and tailored directives to legislate for the safe manufacturing and use of these nascent and complex medicines. In Europe, they are classified as Advanced Therapy Medicinal Products (ATMP). In the USA, this is the remit of the Center for Biologics Evaluation and Research (FDA).
Developers of such pioneering biomedicines need to rest assured that, although challenging, there are pathways and support in place to guide them from the initial R&D stages towards clinical trials and commercialisation. It would be helpful to imagine the development of an advanced medicinal product more as an engineering endeavour. It then becomes apparent how each and every component entering the manufacturing would need to be defined, qualified and controlled at origin, in-process and at release.
What exactly are those components? If we look at gene therapy, the medicinal product consists of or contains recombinant nucleic acid sequences or genetically modified organisms, viruses or cells. In all those cases the components are the starting materials and reagents (also called raw materials) used in the entire generation of the vectors, viruses, plasmids or genetically modified organisms/cells. Despite a wide diversity of approaches, they all commonly rely on vector technologies in order to deliver the genetic material to cells and produce viral vectors either as a step in the protocol or as the finished product.
It is generally at this point, when the extent and complexity of starting materials is fully taken on board in the translational pipeline, that developers stumble in one crucial aspect: current Good Manufacturing Practices, or cGMP. In fact, to monitor and guarantee their safety, advanced medicines need to follow strict and specific cGMP regulations (which in Europe are detailed especially for ATMP), similarly to what applies to standard pharmaceuticals. And this is a major challenge. What for pharmaceuticals is a tried and tested industry reliant often on biochemical synthesis, for advanced medicines it is the engineering of living systems into a final active product.
In essence, cGMP require that each material used to produce or that has come into contact with the final medicinal product needs to have extensive proof of origin, safety documentation testing for infectious agents, lack of animal components and complete traceability down to the smaller reagent and equipment. Reagents that satisfy these requirements are considered of superior, or pharmaceutical, quality. Such quality not only guarantees patient safety but also cuts the burden of risk assessment to the developers. In fact, while research-grade reagents are not forbidden, they incur bespoke and complex due diligence and risk assessment to receive approval by regulators. Regulators expect and highly recommend pharmaceutical quality with clinical intended use, which is achieved by manufacture in certified GMP facilities.
It is the difference between conducting small-scale studies with research-grade reagents normally used in academia, to running a highly regulated and controlled manufacturing process in cell and gene factory environment.
Of all aspects that concern cGMP, that of the controlled facility remains the most elusive to people looking to transit their discoveries to clinic. However, a basic understanding on their inner workings is extremely useful, as it informs the new environment in which their therapeutic will be built. Everything that enters the cGMP process, from staff to equipment to raw materials, has to negotiate with the limits and constrains of this regulated space.
To date, there is not a harmonised standard as most facilities are bespoke and subjected to national regulation and inspections. As is expected from a fast-developing field, regulations around GMP facilities are constantly amended. Nevertheless, irrespective of design, size and individuality, there are some general, top-level aspects that apply to a cGMP environment.
If we imagine to ‘dive’ into a cGMP facility, this is what we are to expect:
Considering the full scale of transitioning to cGMP environments, researchers would be grateful to adapt early to innovations that can ease the task ahead. As we have seen above, even in the best of scenarios, a gene therapy faces a very complex manufacturing process.
Many aspects are out of the control of developers, but the choice of reagents that are of the highest quality, conforming to cGMP requirements and possibly facilitating scalable production are to be considered. If there is one guarantee, it is that the arena of advanced medicinal products will likely become more regulated in the coming years.
However, a silver lining is that more companies are coming together to support developers of cell and gene therapies with reagents that are of clinical standard. For the first time, we are entering a space where developers of gene therapies as well as viral vector manufacturers can count on reliable alternatives to research-grade reagents to cover their cGMP starting materials needs.
At Polyplus-transfection®, we understand the demanding cGMP requirements of developers as they move towards pre-clinical scalable manufacturing and clinical trials. For this reason, we have worked to develop pharmaceutical grade transfection reagents making the transfection reagent suitable for viral vector manufacturing as well as safe for direct administration in humans, taking into account the expected increase in regulatory stringency which medical device GMP grade reagents may fall short of answering as they are often labelled “for research use only and further manufacturing. Not for use in humans or animals”.
We developed gene therapy reagents gold standard PEIpro®-GMP for adherent and suspension systems and for FectoVIR®-AAV GMP superior and industrial AAV vector production in suspension cells.
PEIpro®-GMP and FectoVIR®-AAV GMP are produced in cGMP accredited facilities and come with full traceability documentation suitable for clinical submission to regulatory authorities in the EU, USA and worldwide. Patient safety is also assured by a chemically defined and animal component free composition, certificate of origin, analysis and extensive quality control documentation.
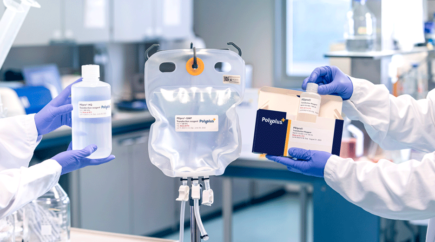
Furthermore, to facilitate large-scale manufacturing in close system ensured by aseptic connections, PEIpro®-GMP and FectoVIR®-AAV GMP come in ready-to-use bags with MPC connectors and weldable tubing.
Find out how our range of highest quality grade PEIpro®-GMP and FectoVIR®-AAV GMP reagents can assist your gene therapy manufacturing needs.
References and resources
(To receive the next articles directly in your mailbox, subscribe to our cGMP VIP list)
PEIpro® is the leading PEI-based DNA transfection reagent that offers flexibility and scalability for viral vector manufacturing.

Available at research and GMP grade to intensify production of recombinant AAV rAAV viral vectors at any scale from benchtop to 2000L scale bioreactor...